









新药审批的唯一注脚
核心提示:帕拉米韦注射液在H7N9禽流感疫情流行之时通过了生产现场检查获批上市,引发行业热议:国家的药品审批机制是否科学有效,直接影响着这个国家的公共卫生安全和药物创新环境,进而影响到整个医药产业发展的效率与走向。
帕拉米韦注射液在H7N9禽流感疫情流行之时通过了生产现场检查获批上市,引发行业热议:国家的药品审批机制是否科学有效,直接影响着这个国家的公共卫生安全和药物创新环境,进而影响到整个医药产业发展的效率与走向。
事实上,该药的安全性和有效性的技术审评已于去年12月由原国家食品药品监督管理局药品审评中心(CDE)完成。4月7日,CDE在官方网站上发布“帕拉米韦注射液”审评概述。长约1700字的论述透露出一个信号:每一个新药的审评结论,都是对这个机构评审能力的一次挑战;在批准一个新药上市前,CDE必须清晰地告知公众,该机构是基于什么数据和实证作出“批”与“不批”的决策的。
临床急需紧扣安全风险
当今,全球药物创新的首要领域就是解决未被满足的临床需求。在药物创新进程中须经历的“临床准入”和“市场准入”两个节点上,CDE发挥着鼓励创新和控制风险并重的作用。而加快新药审批、建立创新药临床研究IND机制,正成为企业最大的心声。
我国自建国以后建立了药品标准管理制度、药品检验制度,于上世纪80年代中期建立了上市药品的审评制度。随着医疗健康事业的发展和医药经济的进步,这一制度在不断发展和完善。以药监体系内部的力量所倡导的审评机制改革已经开展了十多年,以CDE的资源和能力所开展的内部调整和改革在过去的十年中也已进行了4次。
年初《关于深化药品审评审批改革进一步鼓励药物创新的意见(征求意见稿)》进一步指出,完善审评审批机制,加快创新药物审评。为此,《征求意见稿》提出三条措施:一是鼓励以临床价值为导向的药物创新;二是调整创新药物审评技术要求和规范;三是优化审评流程,提高审评工作效率。创新药物研发和审评应当以临床价值为导向,在关注物质基础的新颖性和原创性的同时,更加重视临床价值的评判。
然而,什么样的药物是以临床价值为导向?什么样的药品有可能满足未能满足的医学需求?对此,CDE负责人坦言,通过快速审评通道鼓励创新的新药能否降低药价,这是公众的热切期待;另一方面,企业的创新药批出来以后如果不能进入医保,遭殃的还是企业。CDE对于临床需求的评估体系,决不能抛离于现实环境自我定义一个“临床急需”,审评的决策一定是基于有效性和安全性为基础的效益风险比,这是决定一种药物能否上市的唯一标准。其他任何因素,不论是政治的、宗教的还是经济的,都不是审评员在决策过程中应该考虑的。
据介绍,CDE正加紧与中华医学会在药物流行病学、临床急需用药方面进行一系列合作,旨在将用药需求的信息从临床终端反馈至新药研发上游,了解疾病在中国的发病率和分布情况以及危险因素有哪些,审评机构、工业界和临床界将系统评估新药“临床价值”的内涵,为审评什么样的药品、不审评什么样的药品找到依据。
CDE负责人认为,相较于欧美,药品市场价格机制比较健全,药价因素是由市场、医疗保险来进行调控的。而这些工作,发达国家均有大量的人力、物力投入。
行政审批与技术审评权重
年初发布的另一份“重磅”文件——《2012年中国药品审评报告》(以下简称《报告》)除了展现大量行业为之关切的重点审批品种和数据以外,作为中国药审制度建立以来第一份“白皮书”,并由CDE发布,其背后的政治意义重大。
“长期以来,将药品技术审评等同于行政审批的管理思维制约了我国药审事业的发展,也制约了医药产业的创新发展。” 中国医药工业科研开发促进会执行会长宋瑞霖认为,中国的药品审评人员人数和审评收费不论是种类还是数量都与国际上中等国家的水平相差甚远,审评人员严重短缺,极低的收费导致大批低水平药品申报占据了大量的审评资源, 必须要认真研究探索药品技术审评工作的本质与规律,要将技术审评管理与行政管理区别开来。
宋瑞霖的观点在《报告》中得到印证,《报告》指出,目前我国审评堵塞的核心矛盾在化学药,而化学药的核心矛盾依然在仿制药。我国仿制药申请量高、仿制药重复申请严重、工业化能力不足等问题依然没有得到根本解决。
对此,时任全国人大常委会副委员长的桑国卫在去年“国家创新药物发展政策专家座谈会”上建议,可以采用国际通行做法,通过提高审评收费的方式,进一步规范药品申报,使那些创新性强、临床急需的药品能够尽快得到审评。审批收费标准太低,致使一部分企业随意申报,挤占了许多审评资源。然而有学者指出,将技术审评管理与行政管理区别开来,收费工作也是一样,不能将技术审评收费与行政管理收费混为一谈。不能用收费高低来谈企业负担,低收费基础上的低效率,排队一排3 年,这对企业发展而言实际上是最大的负担。
中国药品注册审批制度的船头正在渐次调整。所有有关创新扶持政策能否到位,也令CDE倍感挑战。如何恰如其分地扮演好自己的角色?如何跟其它角色互补互动交流?如何把引领创新的思考更加准确、清晰、全面地传递给国内外企业机构及其它部委?上述种种问题和挑战丢给的不仅仅是CDE。
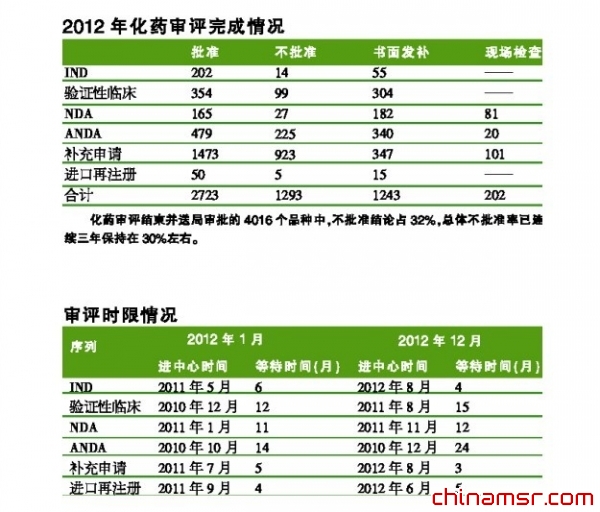
2012年,举CDE之力,力保创新药临床试验申请的审评,使审评等待时间略有缩短并基本维持在4个月左右;上市后补充申请的等待时间也从2012年初的5个月,降至2012年底的3个月;但是,ANDA的等待时间从年初的14个月延长至年底的24个月。同时,NDA和验证性临床试验申请的等待审评时间也有所延长。
2012年国内申请人提出的化药IND申请,大部分审评用时(包括等待时间)在8个月以内(72%),以6~7个月居多(45%),5个月以内占11%,用时超过9个月的品种(15%)多数为复方申请。从治疗领域看,抗肿瘤药物所用时间最短。
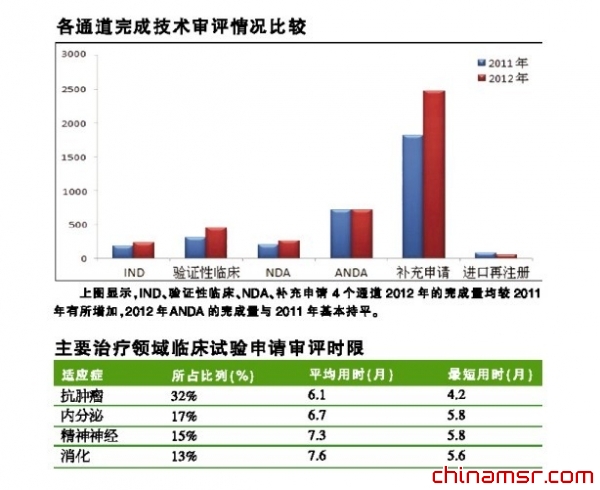
一些原研进口药品对于解决我国未被满足临床需求,提供最新治疗手段发挥着重要作用。药品审评中心关注国内临床亟需的进口药品审评,以使我国公众尽快用到全球最新的药品。通过合理配置审评资源,努力缩短具有重要临床价值的进口药品国内外上市时间的差距。如2012年批准进口上市的苹果酸舒尼替尼胶囊(新适应症)、克唑替尼胶囊、利匹韦林片、替格瑞洛片等,与美国FDA批准上市时间仅间隔一年。
责任编辑:医药零距离
 医院新规:查医药代表 挂钩产品
医院新规:查医药代表 挂钩产品大医院严查医械代表再升级!私下接触医务人员,直接停止采购公司代理产品。...
 编外人员被收保证金?医院取消编制大势所趋
编外人员被收保证金?医院取消编制大势所趋看到一家县级医院向编外人员收取5000元工作保证金,限时不交清者,医院不再使用,老徐认为:编制制度或早已不适应医院发展需要了。...
 三甲医院:8个药询价 要求至少稳定供货半年
三甲医院:8个药询价 要求至少稳定供货半年三甲医院:供货不稳定,踢出一年。...
 5省497名执业药师挂证被查实
5省497名执业药师挂证被查实今年“3•15”后,执业药师“挂证”问题引起全社会关注,按照国家药监局要求,自2019年5月1日起,各省级局组织对行政区域内的药品零售企业开展监督检查。...
 又一大药陷入致癌风波 多家外企全球召回产品!
又一大药陷入致癌风波 多家外企全球召回产品!一些雷尼替丁药物又被查实含有NDMA杂质,目前山德士、葛兰素史克、印度瑞迪博士药厂均已停止雷尼替丁的供应并召回!...
 大洗牌!国务院检查组,进入医疗器械企业了
大洗牌!国务院检查组,进入医疗器械企业了国家严惩在医疗器械购买、销售、纳税...等多个环节的违规情况。...
 4+7全国扩围,中标结果流出
4+7全国扩围,中标结果流出大跌眼镜!4+7全国扩围结果出炉,有外企低价入围,部分原中选药企落标,多个品种再次刷新底价!...
 国务院发文:医械行业,筛选重点企业监管
国务院发文:医械行业,筛选重点企业监管当监管方式越来越科学,不合规或打擦边球的械企面临的压力就越来越大。...
 报告显示:多数医生不再愿意接待医药代表
报告显示:多数医生不再愿意接待医药代表报告显示,医生不再愿意接待医药代表,医药代表的必要性在降低。...
 750家医药企业,最新离职率公布
750家医药企业,最新离职率公布2019年1月到6月的市场薪酬数据白名单公布,其中医药行业的行业增长和薪酬增长都维持稳定的高位;此外,报告采集了750家医药公司的数据,医药人的平均离职率仅为5.91%,为所有行业中最低的。...
 63个药,底价曝光(附名单)
63个药,底价曝光(附名单)(9月6日),山东省药品集中采购网发布《关于山东省药品集中采购拟备案采购产品最低外省及拟挂网价格公示的通知》(以下简称《通知》)。...
 医药业平均月薪公布 仍是最好的就业去向之一
医药业平均月薪公布 仍是最好的就业去向之一据东方财富Choice数据,医药行业平均月薪为1.03万,同比涨幅较大,为11.75%,仅次于公用事业等行业,排名第六名。...